Condividere:



Sintomi, cause e trattamenti della sindrome di Pearson
il Sindrome di Pearson È una delle malattie rare conosciute, a causa della sua bassa prevalenza. Consiste in una malattia di tipo mitocondriale che colpisce l'intero corpo, cioè la sua affettività è multi-sistema. La sua insorgenza si verifica durante l'infanzia e si verifica a causa della cancellazione del DNA mitocondriale.
Questa sindrome fu descritta per la prima volta nel 1979 da Howard Pearson, un pediatra specializzato in ematologia. Un decennio dopo, sono state scoperte le delezioni del DNA mitocondriale che causano questa sindrome.
Cause della sindrome di Pearson
Questa malattia multisistemica è causata da un'anomalia nella fosforilazione ossidativa, che è il processo metabolico mediante il quale l'energia rilasciata dall'ossidazione dei nutrienti viene utilizzata per produrre l'adenosina trifosfato (ATP). L'anormalità di questo processo è dovuta alla duplicazione del DNA mitocondriale.
Nonostante sia una malattia mitocondriale, cioè trasmessa dalla madre, si è concluso che la sindrome di Pearson è di solito sporadica. Pertanto, vi sono delezioni di DNA mitocondriale che fungono da criteri diagnostici, ma la distribuzione casuale di questo tipo di DNA fa convergere le cellule normali e altre con mutazioni.
Questo fatto, chiamato eteroplasmia, che si verifica quando un individuo presenta una miscela di diverse popolazioni di mitocondri, è la causa della grande variabilità nell'espressione clinica della malattia. Questo termine si riferisce al fatto che, nonostante la risposta alla stessa diagnosi, diversi individui mostreranno sintomi diversi, oltre a diversi livelli di affettività.
Qual è la sua prevalenza?
Essendo una malattia rara, colpisce una minoranza della popolazione. Secondo il portale europeo di Malattie Rare, Orphanet, la sindrome di Pearson ha una prevalenza di <1 / 1.000.000.
Inoltre, aggiunge che non ci sono più di 60 casi descritti. Il tipo di eredità trasmessa dalla sindrome di Pearson, in quanto non correlata al sesso, colpisce allo stesso modo sia i ragazzi che le ragazze.
Quali sono i suoi sintomi?
L'esordio della sindrome di Pearson è nella fase infantile e ci sono pochi casi descritti che riguardano neonati. I primi segni sono visibili durante il periodo dell'allattamento e prima dei sei mesi di vita.
Questa sindrome presenta un'immagine molto varia, con condizioni diverse. Ci sono tre caratteristiche che vengono presentate da chiunque soffra della sindrome di Pearson e che sono le seguenti:
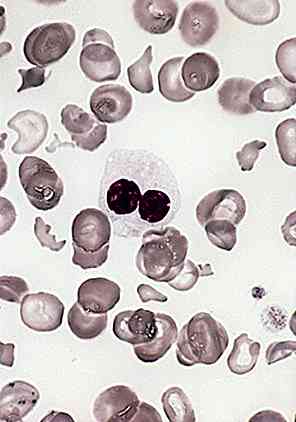
Anemia sideroblastica refrattaria
È il sintomo per eccellenza della sindrome di Pearson e comporta l'alterazione della sintesi dell'emoglobina nei precursori del midollo osseo. In questo modo vengono prodotti i cosiddetti sideroblasti ad anello.
Per il suo trattamento, è conveniente controllare l'anemia e, inoltre, per prevenire il sovraccarico di ferro.
A volte, questa anemia è associata a una neutropenia profonda che consiste nella diminuzione del numero di neutrofili (comunemente noti come leucociti o globuli bianchi).
Anche di thrombocytopenia; quando c'è una situazione ematologica anormale e il numero di piastrine è inferiore. Si verifica a causa della distruzione del tessuto eritrocitario nel midollo osseo.
Vacuolizzazione dei precursori del midollo osseo
Le cellule che sono i precursori del midollo osseo, nel caso della sindrome di Pearson, aumentano considerevolmente le loro dimensioni.
Disfunzione esocrina del pancreas
Questa disfunzione è l'incapacità del pancreas esocrino di svolgere le funzioni digestive in modo normale. Di solito è prodotto da un'improvvisa riduzione della secrezione pancreatica. È strettamente correlato alla cattiva digestione e, di conseguenza, porta al malassorbimento di alimenti non digeriti che spesso innescano uno stato di malnutrizione.
Esiste una grande variabilità nell'espressione della sindrome di Pearson, dovuta al fatto che le cellule patogene sono insieme a quelle normali. Perché una persona presenti manifestazioni patologiche, deve accumulare una quantità sufficiente di DNA mutato. A volte, a causa dei diversi organi e sistemi che sono interessati, si ritiene che la sindrome di Pearson sia un'associazione "incoerente" di sintomi.
In una pubblicazione del Ospedale Universitario Doce de Octubre di Madrid, che consisteva in uno studio di tre casi di sindrome di Pearson, rivelano altri sintomi che di solito si verificano dopo sono oculare, endocrino, cardiaco e condizioni neurologiche. Per quanto riguarda le condizioni cardiache, alcuni pazienti hanno richiesto l'impianto di un pacemaker.
In misura minore, ci sono pazienti con diagnosi di sindrome di Pearson che hanno alterazioni del cervello e / o del tronco cerebrale visibili attraverso la risonanza magnetica.
Inoltre, alcuni di essi presentano l'hiperlactatorraquia, noto anche come ipoglucorraia che suppone la diminuzione dei livelli di glucosio nel liquido cerebrospinale.Inoltre, iperproteinorraquia, aumento di proteine nel liquido cerebrospinale e diminuzione di acido folico in questo liquido sono comuni.
Come si può diagnosticare la sindrome di Pearson?
Normalmente la diagnosi può essere fatta sulla base dei sintomi osservati. Tuttavia, e come indicato dalla Pearson Syndrome Association, è necessario eseguire diversi test ed esami per concludere nella diagnosi di questa sindrome.
In primo luogo, si sospetta una sindrome mitocondriale, può essere effettuata un'analisi preventiva per determinare le più frequenti alterazioni genetiche nel DNA mitocondriale.
Un altro test molto importante nella Sindrome di Pearson è la biopsia muscolare e nel caso in cui i diversi sintomi si uniscono, è essenziale. Questo test prevede la rimozione di un piccolo campione di tessuto muscolare da esaminare e analizzare. È un test rapido e minimamente invasivo e non è nemmeno doloroso.
La neuroradiologia è utile nella diagnosi di questa sindrome poiché offre immagini dello stato del cervello e sarà possibile rilevare l'esistenza di qualche anomalia. Grazie agli studi di laboratorio, i livelli di acido lattico e liquido cerebrospinale può essere misurata e quindi stabilire se coi mezzi, se ci sono dei livelli anormalità.
Ultimo ma non meno importante, vengono effettuati test che analizzano l'attività degli enzimi.
Nei casi in cui vi siano sintomi cardiaci o che colpiscano altri organi o sistemi, come la visione, verranno eseguiti i test corrispondenti per applicare il trattamento richiesto. Gli studi gastroenterologici e nutrizionali possono anche essere eseguiti per verificare che l'assorbimento dei nutrienti venga eseguito correttamente.
trattamento
Ad oggi, la sindrome di Pearson richiede un trattamento sintomatico. Cioè, non ci sono terapie o farmaci per curare la malattia e, quindi, i trattamenti mirano ad alleviare i sintomi che questa sindrome provoca nelle persone che ne soffrono.
A tal fine e, in primo luogo, è molto importante aver effettuato un'analisi esaustiva fornisce i dati sullo stato di salute del bambino e quali sono i loro punti deboli di concentrarsi sul modo più appropriato di trattamento. Inoltre, sono necessari controlli medici per verificare l'evoluzione e verificare che il trattamento utilizzato sia appropriato.
Normalmente, il trattamento sarà mirato ad alleviare episodi infettivi e problemi metabolici.
Nei casi in cui l'anemia è grave, verranno prescritte trasfusioni di sangue. In alcuni casi, tale trattamento sarà accompagnata da terapia con eritropoietina consistente nell'applicare un ormone che contribuisce alla creazione di globuli rossi noto anche come eritrociti.
Inoltre, se esistono, disturbi endocrini o sintomi che colpiscono altri organi che non sono state menzionate in questa sezione saranno discussi e di cui sopra, come il sistema visivo, cuore, ecc
È mortale?
Sfortunatamente, la sindrome di Pearson di solito finisce la vita di questi bambini prima dei tre anni. Le cause sono varie e, tra queste, ci sono:
- Rischio di sepsi che è la risposta massiccia del corpo a un processo infettivo.
- Crisi metaboliche con acidosi lattica o insufficienza epatocellulare.
Non ci sono dati che ci diano informazioni sul tasso di sopravvivenza dei bambini affetti da questa sindrome. Ma se questi bambini a sopravvivere i sintomi, la sindrome di Pearson scompare a causa di evoluzione fenotipica, i sintomi scompaiono spontaneamente ematologiche.
Per quanto riguarda i segni neurologici e miopatici possono aumentare o scomparire. In alcuni casi, la sindrome di Pearson risulta in un'altra malattia mitocondriale che è la sindrome di Kearns-Sayre.
Qual è la sindrome di Kearns-Sayre?
Questa sindrome, anche di tipo mitocondriale, oftalmoplegia progressiva esterna (progressiva debolezza dei muscoli oculari e solleva la palpebra), retinite pigmentosa (gruppo di malattie degenerative dell'occhio) e la sua insorgenza è prima dei 20 anni è caratterizzata. Alcune caratteristiche comuni aggiuntive includono sordità, atassia cerebellare e blocco cardiaco.
Le cifre sulla sua prevalenza offerte da Orphanet stimano che la sindrome di Kearns-Sayre colpisce una persona su 125.000.
La malattia di solito si verifica durante l'infanzia con i seguenti sintomi: ptosi (distacco totale o parziale di un organo), retinite pigmentosa e oftalmoplegia esterna progressiva. Successivamente, altri sintomi compaiono in base alla distribuzione dell'anomalia molecolare, come nella sindrome di Pearson.
Altri sintomi associati a questa sindrome sono la sordità neurosensoriale bilaterale, affettazioni cardiaci, gli effetti del sistema nervoso centrale (atassia cerebellare, disartria, debolezza facciale bilaterale, ritardo mentale), miopatia di disturbi muscolo scheletrico, intestinali ed endocrine (pubertà ritardata , ipoparatiroidismo, diabete) e insufficienza renale. La progressione della malattia è lenta e può durare fino a decenni. Durante questi anni possono apparire nuovi sintomi o peggiorare quelli che sono già presenti.
La sindrome di Kearns-Sayre è anche causata dalla delezione di frammenti di DNA mitocondriale, che influenza il processo di fosforilazione ossidativa. Ci sono casi eccezionali di questa sindrome che si verificano senza la delezione del DNA mitocondriale e sono conseguenza di mutazioni puntiformi localizzate al suo interno.
La diagnosi viene solitamente fatta sulla base delle manifestazioni e, successivamente, dei test effettuati per confermarla. I test sono generalmente gli stessi del caso della sindrome di Pearson. Normalmente la diagnosi non viene fatta nella fase prenatale.
La maggior parte dei casi di questa sindrome si verificano sporadicamente. Le delezioni del DNA mitocondriale vengono trasmesse da una generazione all'altra, eccezionalmente. Si stima che meno del 4% delle donne trasmette la loro prole alla soppressione del DNA mitocondriale. Nel caso degli uomini, non lo trasmettono.
Allo stesso modo, il trattamento di questa sindrome cerca di alleviare i sintomi causati. Si raccomandano controlli regolari da parte di un cardiologo. Nei casi in cui si verificano blocchi cardiaci, richiederanno l'impianto del pacemaker o un dispositivo che defibrillatore per migliorare la qualità della vita di questi pazienti.
I pazienti non udenti possono usare apparecchi acustici. Inoltre, il supplemento al coenzima Q10 si è rivelato utile in alcuni casi. Nel caso di manifestazioni oftalmiche, possono essere trattati chirurgicamente, anche se il rischio di recidiva e le possibili complicanze oculari è alta.
La prognosi della persona che soffre della sindrome di Kearns-Sayre dipenderà dagli organi che hanno colpito e dal grado di coinvolgimento in ciascuno di essi. Questo fatto è strettamente correlato alla proporzione di DNA mitocondriale influenzato e sano che si trova in ciascuno di essi.
In un gran numero di casi, l'aspettativa di vita delle persone che soffrono di questa sindrome può essere normale se ricevono un'adeguata assistenza medica, seguendo il trattamento e le linee guida prescritte dagli operatori sanitari.
bibliografia
- McShane, M.A. (1991) Sindrome di Pearson e encefalomiopatia mitocondriale in un paziente con una delezione di mtDNA. Dipartimento di Neurologia, Ospedale per bambini malati, Queen Square, Londra.
- Sindrome di Kearns-Sayre. Orphanet (2014).
- Sindrome di Pearson. Orphanet (2006).
- Cánovas, R. de la Prieta, J.J. Alonso, C. Ruiz, T. Pereira, C. Aguirre. Anemie sideroblastiche (2001). Servizio e presidente di medicina interna. UPV / EHU. Cruces Hospital. Barakaldo.
- Martín Hernández, M.T. García Silva, P. Quijada Fraile, A. Martínez de Aragón, A. Cabello, M.Á. Martin. Sindromi Pearson e Kearns-Sayre: due malattie mitocondriali multisistemiche, dovute a delezioni nel DNA mitocondriale (2010).
- Cammarata-Scalisi, F., Lopez-Garcia, E., Imperatore, S., Ruiz-Pesini, E., Da Silva, G., Camacho, N., Montoya, J. Sindrome di Pearson. Rapporto di un caso (2011).