Condividere:



Sintomi, cause e trattamento della sindrome di Smith-Lemli-Opitz
il Sindrome di Smith-Lemli-Opitz (SLO) è un disordine metabolico che comprende diversi sintomi, come crescita significativamente lenta, tratti caratteristici facili, microcefalia (dimensioni della testa più piccole del normale), ritardo mentale che può essere lieve o moderato, difficoltà di apprendimento e problemi di comportamento.
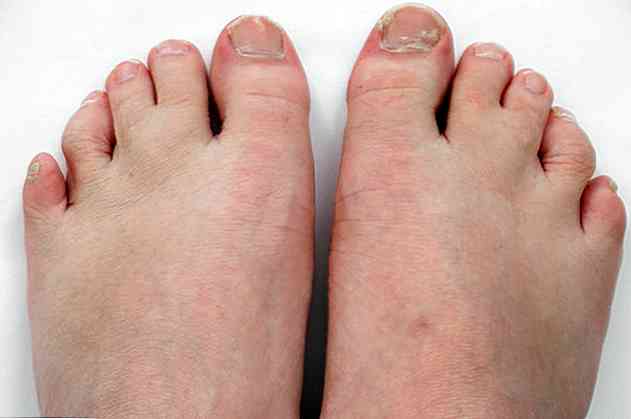
Inoltre è accompagnato da malformazioni nei polmoni, nel cuore, nei reni, nell'intestino e persino nei genitali. Inoltre, possono presentare sindattilia o fusione di alcune dita o polidattilia; il che significa che hanno più di 5 dita su un piede o una mano.
Sembra essere dovuto alla mancanza di un enzima che è importante per metabolizzare il colesterolo che viene acquisito dall'eredità genetica del modello autosomico recessivo.
Tuttavia, tali presentazioni sembrano variare molto a seconda della gravità della malattia anche nella stessa famiglia.
Questa sindrome può comparire in letteratura con nomi quali: deficit di 7-deidrocolesterolo reduttasi, sindrome di RSH o sindrome SLO.
Un po 'di storia ...
Nel 1964, i pediatri David Smith, Luc Lemli e Opitz John descrissero 3 pazienti maschi con microcefalia e ipogenitalismo e definirono questa condizione come RSH dalle iniziali dei cognomi originali di questi pazienti.
Successivamente, il nome della sindrome fu cambiato con i cognomi degli scopritori (SLO).
Circa 30 anni dopo, Tint et al. (1994) scoperto in 5 pazienti con questa condizione, concentrazioni di colesterolo nel sangue significativamente basse, ma con un aumento di oltre 1000 volte i livelli di 7-deidrocolesterolo. Hanno visto che questo aumento era dovuto alla mancanza di un enzima che dovrebbe trasformare 7-deidrocolesterolo in colesterolo.
Successivamente, è stato identificato il gene DHCR7 associato a questa malattia, la clonazione nel 1998 (Witsch-Baumgartner & Lanthaler, 2015).
statistica
La sindrome Smith-Lemli-Opitz colpisce circa 1 su 20.000 a 60.000 nati vivi in tutto il mondo. Può essere effettivamente ereditato in 1 su 1590-13500 individui, ma questa cifra non viene utilizzata perché molti feti con questa condizione muoiono prima di nascere (Organizzazione Nazionale per i Disturbi Rari, 2016).
Riguardo al sesso, colpisce allo stesso modo uomini e donne, sebbene sia più facilmente diagnosticato nei maschi poiché le malformazioni genitali sono più visibili che nelle donne. Inoltre, sembra che abbia più frequenza nelle persone di discendenza europea; specialmente da paesi appartenenti all'Europa centrale come la Repubblica Ceca o la Slovacchia. Tuttavia, è molto raro nella popolazione dell'Africa o dell'Asia.
Cause della sindrome Smith-Lemli-Opitz
La sindrome SLO appare a causa di mutazioni nel gene DHCR7, presente sul cromosoma 11, che è responsabile per l'invio di ordini per la produzione dell'enzima 7-deidrocolesterolo reduttasi. Questo è l'enzima che modula la produzione di colesterolo e sarebbe assente o molto poco in questa sindrome, il che porta ad una produzione insufficiente di colesterolo che impedirebbe una crescita normale.
Questo produce un grande impatto poiché il colesterolo è importante nel corpo. Consiste in un lipide simile al grasso ottenuto principalmente da alimenti di origine animale, come tuorli d'uovo, latticini, carne, pollame e pesce.
È essenziale che l'embrione si sviluppi senza difficoltà, avendo funzioni importanti come il contributo alla struttura delle membrane delle cellule e della mielina (sostanza che copre le cellule del cervello). Serve anche a produrre ormoni e acidi digestivi.
La mancanza dell'enzima 7-deidrocolesterolo reduttasi causa l'accumulo di componenti nel corpo che possono essere tossici per il colesterolo. Quindi abbiamo, da un lato, bassi livelli di colesterolo e allo stesso tempo l'accumulo di sostanze che possono essere tossiche per l'organismo; causando mancanza di crescita, ritardo mentale, malformazioni fisiche e problemi negli organi interni.
Tuttavia, non è noto con certezza assoluta come questi problemi associati al colesterolo diano origine alla sintomatologia della sindrome Smith-Lemli-Opitz.
Attualmente, nel gene DHCR7 sono state trovate oltre 130 mutazioni correlate alla sindrome, infatti esiste un database che include tutti i casi descritti di SLO con le sue varianti, fenotipi e genotipi.
Sebbene ci siano così tante mutazioni possibili, la maggior parte dei casi appartiene ai 5 più frequenti e il resto è molto raro (Witsch-Baumgartner & Lanthaler, 2015).
Queste mutazioni nel gene DHCR7 sono ereditate con un pattern autosomico recessivo, questo significa che una persona che presenta la sindrome deve aver ereditato il gene mutato da entrambi i genitori. Se lo ricevi solo da uno dei genitori, non presenterai la malattia; ma potrebbe essere un vettore e trasmetterlo in futuro.
Vi è il 25% di rischio che i due vettori genitori abbiano un figlio colpito, mentre il rischio che il bambino sia un portatore sarebbe del 50% in ogni gravidanza.D'altra parte, nel 25% dei casi può nascere senza queste mutazioni genetiche o essere un portatore; tutti questi dati sono indipendenti dal sesso del bambino (National Organization for Rare Disorders, 2016).
Tieni presente che esiste una maggiore probabilità di avere figli con qualsiasi disturbo genetico recessivo se i genitori sono parenti stretti (o consanguinei) rispetto ai genitori che non hanno questi collegamenti.
Che sintomi ha?
I sintomi variano a seconda della persona interessata, a seconda della quantità di colesterolo che possono produrre.
Secondo Jiménez Ramírez et al. (2001), le caratteristiche cliniche coprono diversi aspetti e possono essere molto diverse. Generalmente, si trovano sul viso, sugli arti e sui genitali; sebbene possano coinvolgere altri sistemi corporei.
Molti di quelli affetti hanno caratteristiche tipiche dell'autismo, che influenzano l'interazione sociale. Se la condizione è lieve, si possono vedere solo alcuni problemi di apprendimento e comportamento; Ma nei casi più gravi, la persona può avere una grande disabilità intellettiva e anomalie fisiche che possono portare alla morte.
Ci sono sintomi che potrebbero già essere presenti dalla nascita dell'individuo, sebbene includeremo quelli che si verificano in tutte le fasi della vita:
In più del 50% dei pazienti:
- Mancanza di sviluppo fisico osservato dopo la nascita.
- Ritardo mentale (100%).
- Microcefalia (90%).
- Sindattilia o fusione di 2 o 3 dita (<95%).
- ptosi palpebrale, cioè con una delle palpebre superiori cadute (70%).
- Il meato urinario si trova in un posto diverso dal normale nei maschi, come potrebbe essere nella parte inferiore del glande, tronco o unione tra scroto e pene. È presente nel 70% dei casi.
- Palatoschisi, che si manifesta come una sorta di foro allungato nel palato (50%).
- Mascella molto ridotta o micrognazia.
- Linguaggio molto piccolo (microglossia).
- Orecchie di impianto basso.
- Naso corto.
- Discesa incompleta di uno o di entrambi i testicoli.
- Ipotonia o tono muscolare basso.
- Disturbi alimentari
- Disturbi comportamentali: comportamenti antisociali, autodistruttivi e violenti. I comportamenti di auto-stimolazione autistici appaiono anche come movimenti di bilanciamento ripetitivi.
- Autismo
Dal 10 al 50% dei casi:
- Prime cataratte.
- Polidattilia o un altro dito dopo il mignolo.
- Ritardo nella crescita nella fase fetale.
- Genitali ambigui.
- Difetti cardiaci.
- Rene multicistico.
- Assenza di un rene o entrambi alla nascita.
- Malattie epatiche.
- Iperplasia surrenale
- Anomalie polmonari.
- Eccesso di sudorazione.
- Anomalie cerebrali nelle strutture situate nella linea mediana, come lo sviluppo incompleto del corpo calloso, del setto e del verme cerebellare.
- Acrocianosi: vasocostrizione cutanea che causa un colore bluastro nelle mani e nei piedi.
- Piedi equinoviani.
- Stenosi pilorica (15%)
- Malattia di Hirschprung, che causa una mancanza di motilità intestinale (15%)
- Fotosensibilità.
Altri sintomi:
- Obesità o coma.
- Accumulo di liquido nel corpo del feto.
-Alterazioni nello sviluppo neurologico.
- Problemi neuropsichiatrici, che appaiono più frequentemente quando raggiungono l'età adulta.
- Insufficienza respiratoria a causa di problemi polmonari.
- perdita dell'udito
- Alterazioni nella visione, che possono essere accompagnate da strabismo.
- Vomito.
- costipazione
- Convulsioni.
Come può essere diagnosticato?
Questa sindrome appare dal concepimento anche quando il bambino nasce, i sintomi non sono molto chiari e sono più sottili che nella tarda infanzia o nell'età adulta; soprattutto se sono forme più lievi della malattia. Per questo motivo, viene rilevato in ritardo più volte.
In ogni caso, la cosa più comune è che questa condizione è già sospettata poco dopo essere nata a causa delle malformazioni che solitamente presenta (Steiner, 2015).
Secondo la National Organization for Rare Disorders (2016) la diagnosi si basa su esami fisici e un esame del sangue che rileva i livelli di colesterolo. È essenziale che il bambino venga valutato in tutti i possibili aspetti associati alla malattia come occhi, orecchie, cuore, muscoli scheletrici, genitali e disturbi gastrointestinali.
Come per i test del sangue, un soggetto con SLO avrà una elevata concentrazione di 7-deidrocolesterolo (7-DHC) sangue (precursore essere trasformate reduttasi 7-deidrocolesterolo per il colesterolo), e livelli ben Basso contenuto di colesterolo
Può anche essere rilevato prima della nascita attraverso una tecnica ad ultrasuoni o ad ultrasuoni, un apparato che utilizza le onde sonore per esaminare l'interno dell'utero della donna incinta. Con questa tecnica, è possibile osservare le deformità fisiche di questa sindrome.
Un altro test è l'amniocentesi, che comporta l'estrazione di un piccolo campione di liquido amniotico (dove si sviluppa il feto) per rilevare difetti genetici.La stessa informazione può essere ottenuta attraverso il campionamento dei villi coriali (CVS), estraendo un campione di tessuto dalla placenta.
D'altra parte, i test genetici molecolari possono essere utilizzati per la diagnosi prenatale al fine di osservare se ci sono mutazioni nel gene DHCR7 e se presenterà la malattia o sarà solo un portatore.
Qual è il decorso della malattia?
Sfortunatamente, la maggior parte dei casi più gravi di SLO muoiono poco dopo la nascita. Se c'è una grave disabilità intellettiva, è difficile per queste persone sviluppare una vita indipendente.
Tuttavia, se si ricevono cure mediche adeguate e una buona dieta, questi pazienti possono condurre una vita normale.
Quali trattamenti ci sono?
Attualmente non esiste un trattamento specifico per la sindrome di Smith-Lemli-Opitz. Questo perché l'origine biochimica della malattia non è conosciuta oggi con assoluta certezza poiché il colesterolo ha diverse funzioni complesse nel metabolismo.
Il trattamento medico per SLO si basa sui problemi specifici che si riscontrano nel bambino affetto ed è meglio intervenire precocemente.
Può essere molto utile ricevere supplementi di colesterolo o aumentare l'assunzione di colesterolo attraverso la dieta, per migliorare il livello di sviluppo e diminuire la fotosensibilità. A volte è combinato con acidi biliari.
Per l'intolleranza al sole, è consigliabile che questi pazienti utilizzino la protezione solare, occhiali da sole e abbigliamento adeguato quando si esce all'aperto.
È stato dimostrato che la fornitura di farmaci come la simvastatina può ridurre la gravità della malattia. Anche se, come il fenotipo clinico si verifica durante la mancanza di colesterolo nell'embriogenesi, deve essere somministrato in quel momento (Witsch-Baumgartner & Lanthaler, 2015).
D'altra parte, un farmaco antagonista del precursore del colesterolo tossico che è in eccesso (7-deidrocolesterolo) può anche essere usato per evitare che aumenti. Gli integratori di vitamina E possono aiutare.
Altri tipi di farmaci specifici possono essere utili per sintomi quali vomito, reflusso gastroesofageo o stitichezza.
Chirurgia o parentesi possono essere necessari se ci sono deformità fisiche o problemi muscolari legati a questa sindrome come palatoschisi, cardiopatie, ipotonia muscolare o alterazioni genitali.
In conclusione, è necessario continuare la ricerca su questa sindrome in modo da sviluppare trattamenti più efficaci e specifici.
riferimenti
- Jiménez Ramírez, A; Valdivia Alfaro, R; Hernández González, L .; León Corrales, L .; Machín Valero, Y. e Torrecilla, L. (2001). La sindrome di Smith Lemli Opitz. Presentazione di un caso con diagnosi biochimica. Espirituana Medical Gazette, 3 (3).
- Sindrome di Smith Lemli Opitz. (N.d.). Estratto il 6 luglio 2016 dalla National Organization for Rare Disorders (NORD).
- Sindrome di Smith-Lemli-Opitz. (N.d.). Estratto il 6 luglio 2016 dall'Università dello Utah, Health Sciences.
- Sindrome di Smith-Lemli-Opitz. (N.d.). Estratto il 6 luglio 2016 da Counsyl.
- Sindrome di Smith-Lemli-Opitz. (5 luglio 2016). Estratto da Genetica Home Reference.
- Steiner, R. (1 aprile 2015). Sindrome di Smith-Lemli-Opitz. Ottenuto da Medscape.
- Tint, G.S., Irons, M., Elias, E.R., et al. (1994). Biosintesi difettosa del colesterolo associata alla sindrome Smith-Lemli-Opitz. N Engl J Med, 330: 107-113
- Witsch-Baumgartner, M., & Lanthaler, B. (2015). Compleanno di una sindrome: 50 anni dalla sindrome di Smith-Lemli-Opitz. European Journal of Human Genetics, 23 (3), 277-278.