Condividere:



Sindrome di Horner Sintomi, cause, trattamento
il Sindrome di Horner o sindrome di Bernard-Horner di un disturbo neurologico causato da un'origine di interruzione o lesione delle vie nervose simpatiche da qualche parte nel suo percorso dal sistema nervoso al bulbo oculare (Herrero-Morin et al., 2008).
Clinicamente, la sindrome di Horner è caratterizzata dalla presentazione di diverse alterazioni oftalmologici e simpatici, tra i quali troviamo miosi, ptosi o anhidrosis, tra gli altri (Rodriguez-Sanchez, Vadillo, Herrera-Calo e Morenco de la Fuente , 2016).
La sindrome di Horner può apparire associata ad insorgenza acquisita o congenita. A causa di ciò, la sua eziologia è correlata a un'ampia varietà di fattori: incidenti cerebrovascolari, formazioni tumorali, mal di testa ed emicranie, traumi cranioencefalici, chirurgia, ecc. (Vicente, Canelles, Díaz e Fons, 2014).
Per quanto riguarda la diagnosi, questa patologia richiede sia un esame fisico e oftalmologico, sia l'uso di diversi test. Uno dei test più utilizzati per confermare la sua presenza e individuare la causa della sindrome è il test del collirio, accompagnato da alcune tecniche di neuroimaging (Escrivá e Martínez-Costa, 2013).
Infine, anche se non esiste un approccio terapeutico specifico per la sindrome di Horner, l'obiettivo essenziale dell'intervento medico è il trattamento, il controllo e l'eliminazione della causa eziologica di esso (Mayo Clinic, 2014).
Caratteristiche della sindrome di Horner

La sindrome di Horner è un tipo di patologia che colpisce principalmente l'occhio e le aree circostanti su un lato del viso, a seguito di lesioni a vari rami nervosi (Genetics Home Reference, 2016).
Nello specifico, c'è un'interruzione del percorso simpatico che va dal cervello alle aree oculari (Pizarro et al., 2006).
Il nostro sistema nervoso è diviso in due sezioni in base alle caratteristiche anatomiche di questo (Redolar, 2014):
Da un lato, troviamo il sistema nervoso centrale (SNC), composto principalmente dal cervello o dal cervello e dal midollo spinale.
D'altra parte, il sistema nervoso periferico (SNP) comprende la linfa e nervi cranici spinali responsabili del trasporto tutti i motori e le informazioni sensoriali in modo bidirezionale tra i centri del cervello e le diverse aree del corpo.
Inoltre, in quest'ultima suddivisione, possiamo distinguere altri due sistemi fondamentali:
Il primo si riferisce al sistema nervoso autonomo (ANS), la cui funzione principale è il controllo della regolazione interna del corpo, vale a dire quelle funzioni involontarie o automatiche che sono essenziali negli organi interni.
Mentre l'altro, si riferisce al sistema nervoso somatico (SNSo), responsabile del controllo del flusso di informazioni tra la struttura corporea e gli organi interni, con diverse aree del sistema nervoso centrale.
In quest'ultimo possiamo identificare tre componenti fondamentali, i rami simpatico, parasimpatico e enterico.
In questo caso l'area comprensiva è quella che ci interessa. Questo è essenzialmente responsabile della regolazione della mobilitazione organica e corporale in presenza di un evento o di una situazione di pericolo, reale o potenziale.
Il ramo nervoso simpatico è in grado di controllare un'ampia varietà di movimenti involontari e risposte omeostatiche organiche.
A un livello specifico, è legato alla sudorazione, all'aumento o alla riduzione della frequenza cardiaca, alla dilatazione della pupilla, agli atti di volo motorio, alla broncodilatazione, ecc.
Pertanto, la presenza di lesioni transitorie o permanenti in diverse sezioni del sistema simpatico può causare lo sviluppo delle caratteristiche cliniche della sindrome di Horner.
Questa patologia fu inizialmente descritta dal chirurgo oftalmologico Johann Friedrich Horner (1869) (Ioli, 2002).
Nel suo rapporto clinico, si riferiva a un caso di un paziente di circa 40 anni la cui patologia era caratterizzata da (Ioli, 2002):
- Discesa unilaterale o caduta della palpebra.
- Diminuzione della contrazione pupillare.
- Spostamento del bulbo oculare.
- Alterazione della produzione di sudore.
Inoltre, Horner ha identificato un'associazione significativa di questi risultati clinici con una lesione del nervo simpatico a livello cervicale (Ioli, 2002).
Un numero significativo di casi di sindrome di Horner ha identificato un'associazione di esso con lesioni o blocchi delle fibre nervose simpatiche a vari livelli (Avellanosa, Vera, Morillas, Gredilla e Gilsanz 2006):
- centrale: interruzione localizzata a livello del midollo cervicale, del tronco encefalico o dell'ipotalamo.
- periferica: Livello di interrupt pregangliare localizzato (cervicale mediastino anteriore, ai polmoni o ossa cervicotorácia apice) o post-gangliari (aree del seno cavernoso, base cranica, carotidi o ganglio cervicale superiore).
Pertanto, sindrome di Horner può causare paralisi del muscolo dilatatore dell'iride (miosi), muscolo Müller (ptosi), fibre sudomotorie e vasomotoria (anhidrosis, vasodilatazione, arrossamento, ecc) (Avellanosa, Vera, Morillas, Gredilla e Gilsanz, 2006).
I più recenti classificazioni mediche definiti sindrome di Horner come una condizione medica di carattere neurologico, pregiudizio causato da vie nervose che corrono dal cervello per gli occhi e il viso (Birth Injury Guida, 2016).
Anche se questa è una malattia che di solito non provoca alterazioni significative nella capacità visiva o lo stato funzionale generale della persona interessata, la causa della lesione del nervo può causare altre gravi complicazioni mediche (Genetics Home Reference, 2016).
statistica
Differenti indagini epidemiologiche ritengono che la sindrome di Horner sia una malattia rara nella popolazione generale (Genetics Home Reference, 2016).
In generale, si stima che l'incidenza di questa sindrome sia di circa 1 caso per 6.250 parti (Genetics Home Reference, 2016).
Tuttavia, ci sono pochi dati sulle figure di prevalenza nella fase dell'infanzia, dell'adolescenza o dell'adulto.
Inoltre, l'Istituto Nazionale per i Disturbi Rari (2016) afferma che la sindrome di Horner può verificarsi sia nelle donne che negli uomini, in diverse fasce d'età o in qualsiasi gruppo geografico o etnico / razziale specifico.
Segni e sintomi più frequenti

Le caratteristiche cliniche della sindrome di Horner sono associate all'area oftalmologica e alle funzioni omeostatiche dell'organismo.
Normalmente, tutte le alterazioni di solito si verificano unilateralmente, cioè colpisce solo un lato della faccia o del corpo (National Institute for Rare Disorders, 2016).
Questa malattia è caratterizzata essenzialmente da una triade sintomatica di ptosi, miosi e anhidrosis, che verrà descritto qui di seguito (Pardal Souto Ahi Barbaito, Taboada Perianes, 2014):
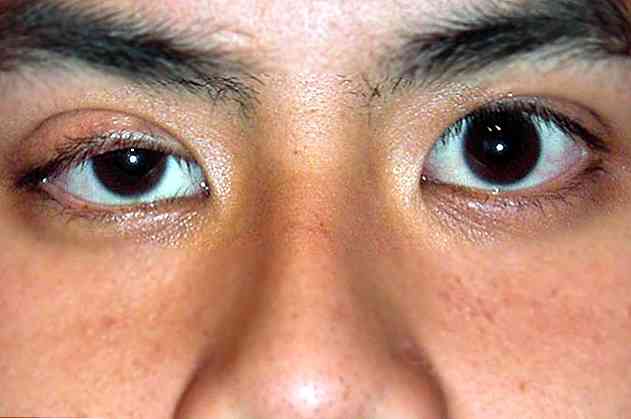
ptosi
Il termine ptosi è usato per riferirsi alla caduta anormale delle palpebre superiori (Institute of Ocular Microsurgery, 2016).
Anche se può essere dovuto a fattori diversi (debolezza muscolare, la pelle cascante, processo di malattia, invecchiamento, ecc), nel caso di sindrome di Horner è causata da danni ai nervi (National Institutes of Health, 2016).
Nello specifico, è stato associato a lesioni localizzate nel terminale nervoso che innervano il muscolo di Müller (Iolli, 2002).
Il muscolo di Müller, noto anche come muscolo palpebrale levator, è il principale responsabile per mantenere le palpebre in una posizione funzionale e consentire il loro movimento volontario.
A livello visivo, possiamo osservare come la palpebra superiore sia sganciata o in posizione più bassa del solito (National Institutes of Health, 2016).
Normalmente, la ptosi colpisce solo un occhio. Di solito non è associato ad altri tipi di patologie oftalmologiche che riducono la capacità visiva della persona che ne soffre.
Tuttavia, sono stati descritti alcuni casi in cui visione offuscata o doppia, aumento della lacrimazione, episodi di dolore o l'ambliopia (occhio pigro), secondarie di ptosi (National Institutes of Health, 2016) sviluppa.
miosi
Un altro dei segni caratteristici della sindrome di Horner è la presenza di una contrazione anormale dell'iride oculare (Iolli, 2002).
L'iride è una delle strutture degli occhi. È una membrana muscolare che è responsabile, insieme con l'allievo, di regolare la quantità di luce che vi accede attraverso la sua contrazione e dilatazione (National Institutes of Health, 2016).
A livello visivo, identifichiamo l'iride come l'area circolare colorata degli occhi (National Institutes of Health, 2016).
Il più comune nella sindrome di Horner è una disfunzione dei muscoli che controllano l'apertura dell'iride e della pupilla, quindi di solito sembra più chiusa del solito alla stimolazione della luce (Iolli, 2002).
Inoltre, è anche probabile che verranno sviluppati altri tipi di alterazioni (Iolli, 2002):
- Congestione congiuntivale: in molti casi si possono apprezzare l'infiammazione e il rossore dei tessuti oculari congiuntivali.
- Eterocromia dell'iride: si riferisce alla presenza di un colore asimmetrico dell'iride degli occhi, cioè, uno ha un certo colore e l'altro tende ad avere un aspetto grigio o bluastro.
- enoftalmo: con questo termine ci riferiamo a uno spostamento degli occhi. A livello visivo, possiamo osservare come uno o entrambi gli occhi si muovono verso l'interno dell'orbita.
Sebbene non vengano prese in considerazione condizioni mediche gravi, in alcuni casi può compromettere la capacità e l'efficienza visiva della persona interessata.
anhidrosis
Il termine anidrosi è usato nella letteratura medica per indicare un'alterazione della produzione di sudore (National Institutes of Health, 2015).
Nel caso della sindrome di Horner, un'assenza o una drastica riduzione della sudorazione nelle regioni facciali, il collo o le aree toraciche viene solitamente identificato (Iolli, 2002).
Tuttavia, come nei casi precedenti, questa patologia di solito si verifica unilateralmente, interessando un lato della faccia o del corpo (Escrivá e Martínez-Costa, 2013).
Sebbene nei casi lievi non comporti complicazioni mediche significative, l'anidrosi può evolvere verso una significativa alterazione della regolazione della temperatura corporea (National Institutes of Health, 2015).
Altre modifiche
A seconda della gravità delle lesioni nervose, altre complicazioni mediche possono verificarsi, come un generale aumento della temperatura corporea delle aree colpite, rossore al viso, secrezioni nasali, super-sensibilità, lacrimazione (lacrimazione abbondante), tra gli altri ( Iolli, 2002).
cause
La sindrome di Horner può avere un'origine acquisita (dopo la nascita) o congenita (prima della nascita), entrambe correlate a alterazioni neurologiche.
Ci sono una grande varietà di fattori che possono portare a lesioni del ramo nervoso simpatico e, di conseguenza, allo sviluppo della sindrome di Horner.
Normalmente, le lesioni sono solitamente divise in tre gruppi (Mayo Clinic, 2014):
Primo ordine
Il coinvolgimento si trova solitamente nella via nervosa che va dall'ipotalamo, il tronco cerebrale alle aree superiori del midollo spinale.
In questo caso, i fattori eziologici più comuni includono:
- Incidente cerebrovascolare.
- Trauma cranio-encefalico.
- Traumatismo al collo.
- Formazione di tumori
- Patologia o malattia demielinizzante.
- Sirigomelia (formazione di cisti midollari)
Secondo ordine
Il coinvolgimento si trova solitamente nella via nervosa che va dalle aree del midollo spinale alla parte superiore del torace e del collo.
In questo caso, i fattori eziologici più comuni includono:
- Tumori polmonari
- Tumori legati alla mielina (Shwannoma).
- Lesioni aortiche
- Chirurgia toracica
Terzo ordine
L'interessamento è di solito localizzato nelle vie nervose che si estendono dal collo alla pelle del viso e alla struttura muscolare dell'iride e delle palpebre.
In questo caso, i fattori eziologici più comuni includono:
- Lesione aortica nel collo.
- Lesione della vena giugulare nel collo.
- Formazione di tumori o processo infettivo in aree vicine alla base del cranio.
- Episodi di emicrania.
- Episodi di cefalea a grappolo.
D'altra parte, sono stati identificati diversi fattori eziologici che sono più comuni nei bambini (Mayo Clinic, 2014):
- Lesione traumatica al collo o alle spalle durante il processo di nascita.
- Alterazione aortica congenita
- Formazione di tumori a livello neurologico.
VALUTAZIONE
La diagnosi della sindrome di Horner si basa fondamentalmente sull'identificazione dei segni clinici, della lesione nervosa e della causa eziologica.
Analisi clinica
Di solito viene eseguito un esame fisico generalizzato, accompagnato da un'analisi della posizione palpebrale, dell'integrità muscolare dell'iride e della sudorazione.
Nel caso della posizione palpebrale, è possibile identificare i difetti di posizionamento a livello visivo.
Tuttavia, nel caso della contrazione dell'iride e della pupilla, è necessario utilizzare altri metodi oltre all'analisi visiva (Iolli, 2002).
- Stimolazione luminosa
- Prova di colliri
- Test di cocaina cloridrato.
- Test idrossilaminico
Infine, per valutare la sudorazione, viene solitamente utilizzata una registrazione del tasso di produzione di sudore in varie aree del corpo.
Identificazione della lesione del nervo e causa eziologica
In questo caso, le tecniche utilizzate sono fondamentalmente basate sul neuroimaging delle aree cerebrali e periferiche (Iolli, 2002).
- Tomografia computerizzata
- Risonanza magnetica nucleare.
trattamento
Come abbiamo notato nella descrizione iniziale, non esiste una cura o un trattamento specifico per la sindrome di Horner (Mayo Clinic, 2014).
Tutti gli interventi medici si concentrano sul trattamento della causa eziologica (Mayo Clinic, 2014).
Il più delle volte, la sindrome di Horner è dovuta alla presenza di un tumore o di una lesione traumatica. In entrambi i casi, è possibile utilizzare procedure chirurgiche e farmacologiche per la loro eliminazione (Organizzazione nazionale per i disturbi rari, 2016).
riferimenti
- Avellanosa, J., Vera, J., Morillas, P., Gredilla, E., & Gilsanz, F. (2006). Sindrome di Horner e blocco del plesso brachiale omolaterale in caso di analgesia epidurale per il travaglio. Rev. Soc. Esp. Pain.
- Guida per lesioni alla nascita. (2016). Sindrome di Horner Estratto dalla guida per lesioni alla nascita.
- Escrivá, E., & Martínez-Costa, L. (2013). Sindrome di Horner incompleta come segno di presentazione di ependimoma del quarto ventricolo. Arch Soc Esp Oftalmol.
- Herrero-Morín et al.,. (2008). Sindrome di Horner congenita. Anpedi.
- Ioli, P. (2002).La sindrome di Claude Bernard-Horner e altri per sempre estranei. Rivista dell'ospedale della comunità privata.
- Mayo Clinic (2014). Sindrome di Horner. Ottenuto dalla Mayo Clinic.
- NIH. (2015). Palpebre cadute Ottenuto da MedlinePlus.
- NIH. (2016). Sindrome di Horner. Estratto da Genetica Home Reference.
- NORD. (2016). Sindrome di Horner Estratto dalla National Organization for Rare Disorders.
- Pizarro, M., Campos, V., Irarrázaval, S., Mesa, T., Escobar, R., e Hernández, M. (2006). Sindrome di Horner pediatrico: analisi di 5 casi. Ottenuto da Scielo.
- Rodríguez-Sánchez, E., Vadillo, J., Herrera-Calo, P., e Marenco de la Fuente, M. (2016). Sindrome di Horner dopo analgesia epidurale per parto. Rapporto di tre casi. Rev Colomb Anestesiolo.
- Vicente, P., Canelles, E., Diaz, A., & Fons, A. (2014). Sindrome di Horner irreversibile dopo simpatectomia toracoscopica bilaterale. Arch Soc Esp Oftalmol.