Condividere:



Sindrome di Rokitnasky-Küster-Hauser: sintomi, cause e trattamenti
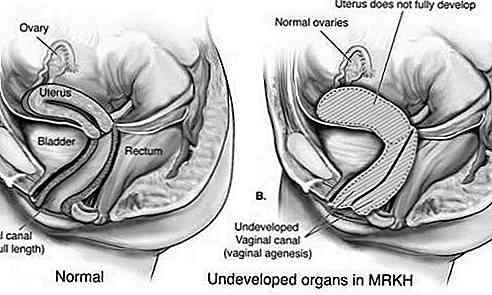
Sindrome di Rokitansky-Küster-Hauser (MRKH) è un disturbo che colpisce il sistema riproduttivo femminile caratterizzato da sottosviluppo o assenza dell'utero e della vagina.
Le donne con questa sindrome sviluppano caratteristiche sessuali secondarie durante la pubertà - ossi, peli pubici - ma non hanno un ciclo mestruale (amenorrea primaria).
Non iniziare il ciclo mestruale è spesso il segno iniziale della sindrome MRKH. Anche se le donne con questa condizione non possono avere gravidanze, se possono avere figli attraverso la riproduzione assistita.
La gravità della sindrome può variare a seconda del tipo. Il tipo 1 è caratterizzato da un'assenza isolata dei due terzi prossimali della vagina. Tipo II è caratterizzata da altri difetti come ad esempio anomalie renali (40% dei casi), anomalie scheletriche (20-25%), perdita di valore (10%) di udito, e, più raramente, difetti cardiaci.
A causa della natura della malattia, è probabile che causi problemi psicologici significativi, quindi è consigliabile chiedere aiuto.
La sindrome MRKH ha un'incidenza mondiale di 1 su 4500 parti di donne, secondo diversi studi.
Di solito viene diagnosticato più frequentemente in adolescenza, quando viene controllato se il ciclo mestruale si sviluppa. Sebbene le donne non possano avere figli, le loro ovaie sono normali e funzionali.
Sintomi della sindrome di Rokitnasky-Küster-Hauser
I sintomi della sindrome MRKH variano molto da una donna all'altra, quindi ricorda che le donne affette potrebbero non avere tutti i sintomi menzionati di seguito.
Sindrome di tipo I di Mayer-Rokitansky-Hauser
Questo tipo è anche noto come aplasia di Müller ed è caratterizzato da uno sviluppo inadeguato dell'utero e della vagina. Nella maggior parte dei casi, l'utero e / o la vagina non si sono sviluppati (aplasia); o c'è un restringimento della parte superiore della vagina e dell'utero (atresia). Anche le tube di Falloppio potrebbero essere colpite.
Alcune delle caratteristiche o dei sintomi della sindrome MRKH sono:
-Prima menopausa o assenza di periodi durante la pubertà. Sebbene questo sintomo sia comune nella sindrome MRKH, il paziente viene sottoposto alla pubertà con normale telarca e adrenarche. Tuttavia, non ci sono le mestruazioni.
Poiché la funzione delle ovaie è normale, i pazienti sperimentano tutti i cambiamenti associati alle mestruazioni o alla pubertà.
-Organismi genitali esterni normali.
- Profondità vaginale ridotta, da 2 a 7 cm.
- Caratteristiche sessuali come seno normale e peli pubici.
-Ovestenti funzionali con normali livelli di estrogeni.
-Normali modelli cromosomici.
-Nella sindrome MRKH tipo 1, solo la vagina e l'utero sono anormali. Nel tipo II MRKH, difetti di vagina e dell'utero possono essere accompagnati da anomalie nel Falloppio, come nei reni o colonna vertebrale.
-Anche se le anomalie cardiache sono rare, possono verificarsi la finestra polmonare dell'aorta, il difetto del setto atriale e la stenosi della valvola polmonare
-I pazienti con sindrome MRKH di solito lamentano dolore addominale ciclico a causa del distacco dell'endometrio ciclico senza una via di drenaggio del brevetto.
-Esterilità: molti pazienti spesso cercano cure cliniche per l'infertilità, ma non per l'amenorrea primaria.
- Un altro sintomo è la difficoltà nei rapporti sessuali, poiché il grado di aplasia vaginale può variare da assenza completa a sviluppo debole, che può causare dispareunia.
-Difficoltà di minzione, incontinenza urinaria, infezioni urinarie ricorrenti.
- Anomalie vertebrali.
-Una fionda di tessuto palpabile può essere presente a livello del riflesso peritoneale.
- Malformazioni renali.
Sintomi della sindrome MRKH Tipo II
L'insufficienza renale è l'anomalia più comune associata alla sindrome MRKH di tipo II.
Le donne con sindrome MRKH tipo II possono avere alcun rene, malformazioni di uno o di entrambi i reni (displasia renale), lo sviluppo di reni (ipoplasia) e / o il posizionamento errato all'interno del corpo di uno o di entrambi i reni (ectopia renale).
Queste anomalie possono causare insufficienza renale crescita, calcoli renali, aumento della suscettibilità alle infezioni del tratto urinario e accumulo anomalo di urina nel rene a causa di ostruzione.
Molte donne con sindrome di tipo II MRKH possono anche avere malformazioni scheletriche come problemi ossei nel vertebre cervicali e toraciche che può svilupparsi in modo improprio.
si possono anche verificare anomalie del volto, come avere un anormalmente piccola ganascia (micrognazia), labiopalatoschisi, e sottosviluppo di un lato della faccia, con conseguente asimmetria facciale.
Molte donne affette possono anche sviluppare problemi di udito, principalmente a causa di anomalie strutturali dell'orecchio medio.
Quando sono coinvolte le orecchie, il disturbo può essere chiamato Sindrome dell'orecchio renale genitale (GRES).
Alcune delle donne con sindrome di tipo II di MRKH hanno avuto altre anomalie fisiche che includono difetti nelle mani e / o nelle braccia.
Queste anomalie degli arti possono includere assenza di uno o più dita e dei piedi, pollice duplicati e assenza di lunga dell'avambraccio sottili (raggio assente) osso. Non tutti i sintomi si verificano in tutti i pazienti, dipende dalla posizione e dalla gravità.
cause
Nella maggior parte dei casi, l'origine della sindrome MRKH è sconosciuta e si verifica in donne senza storia familiare.
I ricercatori indicano i fattori genetici e ambientali come le cause della sindrome, sebbene non siano ancora stati determinati geni o geni associati alla condizione.
Storicamente, i ricercatori hanno suggerito che la sindrome può verificarsi a seguito di malattia o materno esposizione del feto a varie sostanze nocive, come ad esempio alcuni farmaci, abuso di droghe, alcool ...
Tuttavia, non ci sono stati studi che confermino questa associazione tra la sindrome e il consumo di alcuni altri farmaci.
In alcune famiglie, la sindrome sembra avere un modello di ereditarietà dominante. Tuttavia, il modello di ereditarietà è difficile da stabilire, poiché non tutte le donne che ne soffrono hanno gli stessi sintomi, anche se appartengono alla stessa famiglia.
Secondo i ricercatori, è molto probabile una combinazione di fattori genetici e ambientali.
Per quanto riguarda le anomalie nel processo di riproduzione, esse sono dovute a uno sviluppo incompleto del dotto Mülleriano, ma la sua causa rimane sconosciuta.
Sembra che negli ultimi anni ci sia stato un aumento delle prove, in quanto la sindrome MKRH è una malattia genetica. L'aumento dei casi studio ha fatto sì che questa idea continuasse a essere rafforzata.
Stiamo parlando di disordini genetici quando c'è una combinazione di geni per un particolare tratto. Se si tratta di una malattia genetica dominante come serio in questo caso, si sarebbe prodotto dallo sviluppo anormale di una singola copia di un gene anormale.
Questo gene difettoso può essere ereditato sia dalla madre che dal padre o può essere dovuto a una nuova mutazione nel gene stesso.
La probabilità quindi di trasmettere quel gene difettoso sarà del 50% per ogni gravidanza, indipendentemente dal sesso del bambino.
Anche l'ereditarietà poligenica è stata proposta come causa della sindrome MRKH.
Ad oggi, in diverse persone affette dalla sindrome MRKH sono state trovate sette delezioni e una duplicazione di segmenti cromosomici. Ma solo una di queste anomalie è stata trovata per persona.
Attualmente, quei cromosomi sono stati identificati dove è possibile che siano coinvolte le eliminazioni segmentali. Questi cromosomi sono: cromosoma 1 (1q21.1), 4 (4q34q), 8 (8p23, 1), 10 (10p14-15), 16 (16p11.2), 17 (17q12) e 22 (22q11.21) e la duplicazione è stata trovata sul cromosoma X (Xpter-p22.32).
Tutte queste nuove informazioni ha portato i ricercatori a selezionare più geni candidati, tra cui: HNF1B era, LHX1, TBX6, ITIH5 e SHOX.
trattamento
L'obiettivo del trattamento è che il paziente abbia un funzionamento sessuale completo e soddisfacente.
Poiché i sintomi sono così variabili, è necessaria la collaborazione di un team di specialisti, al fine di garantire un approccio globale al trattamento.
Generalmente essi sono incoraggiati quei pazienti con la sindrome di MRKH a cercare aiuto psicologico dopo la diagnosi e prima del trattamento, dal momento che questa sindrome può causare ansia o disagio psicologico. Si consigliano anche gruppi di supporto.
Per quanto riguarda il trattamento dell'aplasia vaginale è quello di creare una nuova vagina.
Il trattamento può essere chirurgico o meno. La non-chirurgica sarà sempre la prima opzione. Una delle opzioni sono dilatatori vaginali, che servono per aiutare a ingrandire o creare una vagina.
Il metodo più conosciuto è il dilatatore Franck, noto anche come dilatatore perineale, che non richiede alcun tipo di intervento chirurgico.
Essere auto-somministrato il paziente deve essere sufficientemente motivato per usarlo. Dura da circa sei settimane a diversi mesi.
Un'altra opzione può essere una vaginoplastica, che creerebbe una vagina poco profonda. Ma su questo intervento non c'è molto accordo su quali tecniche usare.
La tecnica Mclndoe è una delle procedure chirurgiche più utilizzate per la ricostruzione della vagina. Viene prelevato un innesto di pelle prelevato dalle natiche o dalla coscia e applicato a una protesi a forma di pene gonfiabile. Questa protesi con gli stampi per innesto al tunnel vaginale, che si apre con un bisturi e viene dissezionata in modo netto e diretto, con attenzione a non danneggiare il vegija, il retto o il peritoneo.
È lasciato da sette a dieci giorni e quando viene cambiato è sotto anestesia. Successivamente, il paziente utilizzerà altre protesi di dilatazione. A tre mesi il paziente può fare sesso.
Il problema con questa tecnica è che oltre a lasciare cicatrici, è necessaria una dilatazione a lungo termine.
Un'altra tecnica utilizzata per la sindrome MRKH è la neo-vagina intestinale.
Questa tecnica utilizza un segmento isolato dell'intestino. Consiste nel sezionare un frammento del colon sigmoideo attraverso un'incisione addominale e trasferirlo nell'area in cui è stato creato lo spazio della neo-vagina. Questa tecnica è piuttosto complessa e può durare fino a 8 ore.
Il vantaggio di questa tecnica è che lascia una vagina spaziosa di lunghezza adeguata e che è autolubrificante. Per quanto riguarda gli svantaggi, la prima cosa sarebbe che si tratta di un intervento chirurgico complesso e infrabominale, che lascia le secrezioni mucose in eccesso attraverso il frammento intestinale, il prolasso vaginale della mucosa e l'infiammazione della mucosa nella neovagina.
Infine, altre tecniche sono le Tecnica Vecchieti.
Questa tecnica esercita una pressione progressiva continua da un'oliva acrilica attraverso il potere spazio neovaginale e la parete addominale. Quindi un dispositivo di trazione viene inserito nella cavità peritoneale e rimuove gradualmente la volta vaginale. Questa tecnica viene ora eseguita tramite laparoscopia.
riferimenti
- https://visualsonline.cancer.gov/
- https://ghr.nlm.nih.gov/condition/mayer-rokitansky-kuster-hauser-syndrome
- http://emedicine.medscape.com/article/953492-overview
- http://www.news-medical.net/health/Mayer-Rokitansky-Kuster-Hauser-(MRKH)-Syndrome-Symptoms-(Spanish).aspx
- http://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=GB&Expert=3109
- https://rarediseases.org/rare-diseases/mayer-rokitansky-kuster-hauser-syndrome/
- http://www.biomedcentral.com/content/pdf/1750-1172-2-13.pdf
- Immagine sorgente: http://www.mrkhnorge.org/mrkh/?lang=en