Condividere:



Sindrome di Tolosa-Hunt Sintomi, cause, trattamento
il Sindrome di Tolosa-Hunt è un raro tipo di oftalmoplegia dolorosa (Buedo Rubio, Martin-Tamayo Onsurbe Blazquez e Ramirez, 2015).
Clinicamente, questa malattia è caratterizzata dalla presenza di episodi di dolore o preorbitario dell'emicrania, Oculomotor paralisi, alterazioni pupillari e ipoestesia o iperalgesia in varie zone aree (Martinez, Casasco, Pendre, De Bonis e Berner, 2010).
L'origine delle caratteristiche cliniche di questo disturbo è dovuto allo sviluppo di lesioni in vari nervi cranici: III, IV, IV e / o V. provoca conseguenza gonfiore del seno cavernoso, all'apice orbitale e la fessura orbitale superiore (Martinez, Casasco, Pendre, De Bonis e Berner, 2010).
Tuttavia, la causa eziologica di questo processo patologico non è ancora nota esattamente. Solitamente non è correlato a fattori primari o secondari come traumi, tumori, infezioni, ecc. (Granados Reyes, Soriano Redondo e Durán Ferreras, 2012).
Oltre a esame fisico nella diagnosi di sindrome di Tolosa-Hunt solito seguito i criteri proposti dagli International Classificazione mal di testa Malattie Second Edition (Diaz, Aedo e González Hernández, 2009).
In questo caso, il test di scelta per identificare i disturbi nervosi è la risonanza magnetica nucleare (NMR) (Diaz, Aedo e González Hernández, 2009).
Il trattamento standard della sindrome di Tolosa Hunt si basa sulla somministrazione di farmaci per via orale corticoidi (Zimmermann Paiz, 2008).
Caratteristiche della sindrome di Tolosa-Hunt
La sindrome di Tolosa-Hunt è un personaggio oftalmoplegia dolorosa causata da infiammazione idiopatica di varie aree oculari e orbitali (Paiz Zimmermann, 2008).
Il termine oftalmoplegia dolorosa utilizzato in campo medico e sperimentale per riferirsi ad una condizione definita da (Martinez, Casasco, Pendre, De Bonis e Berner, 2010):
- Dolore localizzato nelle regioni orbitali o craniche (specialmente a livello unilaterale).
- Paralisi ipsilaterale dei muscoli oculomotori.
- Anomalie nella contrazione pupillare.
- Alterazioni della sensibilità in varie aree del viso.
Questo quadro clinico è in genere causata da una vasta gamma di processi, tra cui eventi traumatici, malattie vascolari, tumori, processi infettivi o infiammatori, neuropatia diabetica, l'emicrania, ecc (Serralta San Martin, Torrecillas Narvaez, Soler Rangel Gómez Sanz e Ibáñez Cerezo, 2013).
Nel caso della sindrome di Tolosa Hunt, anche se il processo patologico che dà origine al suo decorso clinico non è esattamente conosciuta, le zone più colpite sono Martínez, Casasco, Pendre, De Bonis e Berner, 2010):
- Seno cavernoso: I seni cavernosi si trovano a livello intracranico, circondato dalla dura tra orbitaroio apice e la fessura orbitale superiore. Si tratta di un plesso venoso, cioè, una parete sottile accumulo venoso dover formare una cavità. Al suo interno contiene l'arteria carotide interna e alcune parti craniali (III, IV, V, ecc.).
- Apx orbitale: Si riferisce al canale vertice orbitale attraverso cui l'ottica fibre nervose coroide plesso e l'arteria oftalmica.
- Fessura orbitale superiore: tale struttura si trova nella scanalatura formando la più grande e più piccola zona dello sfenoide (che si trova nella zona interna della faccia in successive limiti delle orbite. Esso contiene il ramo inferiore e superiore del cranica II, IV e VI (muscoli extra oculari innervati).
Questa sindrome è stata descritta per la prima dagli spagnoli neurochirurgo Eduardo Tolosa nel 1954 (Granados Reyes, Soriano Redondo e Durán Ferreras, 2012).
Nella descrizione iniziale, Tolosa si riferiva a un paziente il cui quadro clinico è caratterizzato da:
- Dolore orbitale nell'area sinistra.
- Oftalmoplegia omolaterale.
- Perdita significativa dell'acuità visiva.
- Iposestesia nel primo ramo del nervo trigemino.
L'autopsia ha rivelato un'infiammazione granulomatosa del seno cavernoso (Martinez, Casasco, Pendre, De Bonis e Berner, 2010).
Anni dopo, nel 1982, William Hunt definì un totale di 6 casi simili. Inoltre, è riuscito a dimostrare l'efficacia del trattamento con corticosteroidi nel migliorare i sintomi associati (Martinez, Casasco, Pendre, De Bonis e Berner, 2010).
Smith e Taxal sono stati i primi autori a descrivere questa entità clinica e la sindrome di Tolosa Hunt nel 1966 (Martinez, Casasco, Pendre, De Bonis e Berner, 2010).
Attualmente, criteri clinici e diagnostici per questa sindrome sono stati classificati dalla International Headache Society nel 2004 (Martinez, Casasco, Pendre, De Bonis e Berner, 2010).
È una patologia frequente?
La sindrome Tolosa Hunt è una malattia rara nella popolazione generale (National Organization for Rare Disorders, 2016).
Alcuni autori come Taylor (2015) affermano che è raro negli Stati Uniti e internazionale, anche se esistono dati precisi sulla sua prevalenza sono noti.
record clinici dimostrano Sindrome di Tolosa-Hunt viene a rappresentare circa il 9% delle condizioni infiammatorie orbitali (Zimmermann Paiz, 2008).
Pertanto, si stima che la sua incidenza ammonti a 1-2 casi per milione di persone in tutto il mondo (Aguirre, Zúñiga e Barrera, 2014).
Essa si verifica in modo equivalente in uomini e donne, l'età media di insorgenza 41 anni (National Organization for Rare Disorders, 2016).
Tuttavia, alcuni casi di presentazione precoce in persone sotto i 30 anni sono stati documentati. In alcuni casi sporadici, questa sindrome può colpire bambini di età inferiore ai 10 anni (Organizzazione Nazionale per i Disturbi Rari, 2016).
Segni e sintomi
La sindrome Tolosa Hunt caratterizzata principalmente dalla presenza di una dolorosa oftalmologia (Martinez, Casasco, Pendre, De Bonis e Berner, 2010).
I segni e i sintomi più comuni sono legati alla presenza di episodi di dolore, infiammazione, parestesie, paralisi, ecc.
Infiammazione orbitale
Come notato sopra, uno dei segni caratteristici della sindrome di Tolosa-Hunt è l'infiammazione del seno cavernoso, apice orbitale e la fessura orbitale superiore (Martinez, Casasco, Pendre, De Bonis e Berner, 2010).
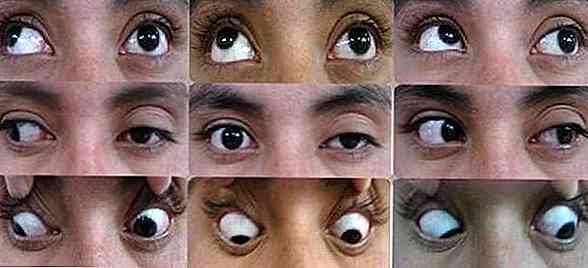
A livello visivo, viene identificata un'infiammazione significativa di tutte le aree facciali che circondano i bulbi oculari (National Organization for Rare Disorders, 2016).
È anche possibile identificare (Organizzazione nazionale per i disturbi rari, 2016):
- proptosis: profusione di bulbi oculari secondari all'infiammazione anatomica delle strutture adiacenti.
- ptosi: flaccidità e abbassamento delle palpebre superiori secondarie a un'affettazione nervosa.
Dolore orbitale
Le persone colpite soffrono spesso episodi ricorrenti di dolore acuto localizzato in orbitali e del viso zone vicino agli occhi (Martinez, Casasco, Pendre, De Bonis e Berner, 2010).
Questo è di solito unilaterale, colpisce un lato del viso, anche se ci sono stati casi con coinvolgimento bilaterale (Granados Reyes, Soriano Redondo e Durán Ferreras, 2012).
È frequente che il dolore si espanda progressivamente verso aree frontali e temporali. Può durare fino a 8 settimane in assenza di un approccio terapeutico efficiente (Martínez, Casasco, Pendre, De Bonis e Berner, 2010).
Inoltre, episodi di dolore sono descritti da persone affette come (Granados Reyes, Soriano Redondo e Durán Ferreras, 2012):
- Intenso.
- Urentes.
- lacerante
- Sharps.
Essa può essere accompagnata da altri sintomi come oculomotor parestesia entro le prime settimane di insorgenza (Martinez, Casasco, Pendre, De Bonis e Berner, 2010).
Questo tipo di dolore deriva fondamentalmente da una nevralgia del ramo oftalmico del nervo cranico V e del ramo mascellare. Tuttavia, l'occupazione può ridurre o eliminare il dolore nelle prime 24 ore (Martínez, Casasco, Pendre, De Bonis e Berner, 2010).
Paresi oculomotoria
Uno dei risultati principali della sindrome di Tolosa Hunt è la presenza di paresi, cioè un'assenza o una significativa diminuzione del movimento volontario.
In questo tipo di patologia, la debolezza muscolare e / o la paralisi parziale influiscono preferenzialmente sulle aree oculomotorie.
Il più comune è che vengono identificati episodi di paresi che influenzano i nervi cranici (Sánchez Iñigo e Navarro González, 2014).
- III (nervo oculomotore): controlla sia la contrazione pupillare che il movimento dei bulbi oculari.
- IV (nervo trocleare): è responsabile del controllo della funzione motoria del muscolo oculare obliquo superiore
- VI (nervi Abducens): è responsabile del controllo delle funzioni motorie oculari esterne, cioè del movimento generato dal muscolo retto laterale.
A livello visivo, possiamo osservare come le persone colpite non sono in grado di eseguire atti motori con gli occhi. Non sono in grado di muovere gli occhi o dirigere lo sguardo verso direzioni diverse (National Organization for Rare Disorders, 2016).
Disturbi pupillari
Nonostante non sia presente in tutti i casi, in alcune persone affette possiamo identificare alterazioni legate alla funzione pupillare.
In questi casi, una delle disfunzioni più comuni è la midriasi o la miosi pupillare.
- midriasi: aumento anormale delle dimensioni e del diametro della pupilla. Si verifica una dilatazione esagerata.
- miosi: diminuzione anormale delle dimensioni e del diametro della pupilla. C'è una contrazione esagerata.
Anomalie visive
Le anomalie oculari e orbitali sopra descritte possono determinare una riduzione variabile dell'acuità visiva (Organizzazione Nazionale per i Disturbi Rari, 2016).
cause
segni e sintomi clinici della sindrome Tolosa Hunt è derivato da non specifico infiammazione dell'apice orbitale, la fessura orbitale e seno cavernoso (Buedo Rubio Martin-Tamayo Blazquez e Onsurbe Ramirez, 2015).
A sua volta, questo processo patologico ha origine in un coinvolgimento dei nervi cranici III, VI, IV e V (Buedo Rubio, Martin-Tamayo Onsurbe Blazquez e Ramirez, 2015).
Tuttavia, la ricerca attuale non ha ancora identificato l'eziologia specifica (Buedo Rubio, Martín-Tamayo Blázquez e Onsurbe Ramírez, 2015).
Oltre a questo, alcuni rapporti clinici indicano i fattori autoimmuni come l'origine della sindrome di Tolosa Hunt (Martínez, Casasco, Pendre, De Bonis e Berner, 2010).
Alcune analisi del siero di coloro che ne sono affetti hanno dimostrato la presenza di anticorpi del lupus, ANCA e antiperossidasi (Martínez, Casasco, Pendre, De Bonis e Berner, 2010).
Inoltre, altre possibili cause sono (National Organization for Rare Disorders, 2016):
- Infiammazione granulomatosa
- Infiammazione generalizzata dei vasi sanguigni cranici.
diagnosi
Come buona parte delle patologie, nella diagnosi della sindrome di Tolosa Hunt, l'analisi della storia clinica familiare e individuale e l'esame obiettivo sono fondamentali per determinare le caratteristiche cliniche del paziente colpito.
Al fine di classificare i segni e i sintomi clinici, vengono generalmente utilizzati i criteri proposti dalla International Classification Headaches Diseases Second Edition (Díaz, Aedo e González Hernández, 2009).
- Uno o più episodi di dolore localizzati nelle aree orbitali a livello unilaterale. Presenta un corso persistente in assenza di cure mediche.
- Paralisi parziale o completa di uno o più dei seguenti nervi cranici: accoppiamento di coppia II, coppia IV, coppia VI o granuloma attraverso biopsia cutanea o risonanza magnetica (MRI).
- Gli episodi di dolore e paresi devono essere risolti dopo 72 ore dall'applicazione di un trattamento adeguato.
- Sono escluse le cause eziologiche associate a tumori, vasculiti, sarcoidosi, emicrania, diabete o meningite basale.
Inoltre, per l'esame preciso di queste caratteristiche, vengono solitamente utilizzati alcuni test di laboratorio.
Nella sindrome di Tolosa Hunt, la risonanza magnetica nucleare (NMR) è la tecnica della lezione per lo studio dell'infiammazione del seno cavernoso (Diáz, Aedo e González Hernández, 2009).
trattamento
Una volta fatta la diagnosi differenziale, il classico approccio terapeutico è la somministrazione di farmaci corticoidi.
Sebbene in alcuni casi sia possibile che vi sia una remissione spontanea dei sintomi. Se non si usa un trattamento con corticosteroidi, questa condizione medica può persistere nel tempo.
La prognosi medica è favorevole. La risoluzione degli episodi di dolore è in genere completa con o senza trattamento specifico, sebbene la remissione sia comune.
Fino al 40% delle persone colpite descrive di aver sofferto vari episodi di dolore orbitale.
Inoltre, in alcuni casi possono persistere alcune alterazioni oculari, specialmente quelle legate alla motilità palpebrale e oculare.
riferimenti
- Aguirre, D., Zúñiga, G., & Barrera, L. (2014). Sindrome da caccia alla tolosa: case report e review della letteratura. Acta Neurol Colomb.
- Buedo Rubio, M., Martín-Tamayo Blázquez, M., e Onsurbe Ramírez, I. (2015). La sindrome di Tolosa-Hunt, una diagnosi di esclusione. Rev Pediatr. Aten. primario.
- Díaz, C., Aedo, I., & González-Hernández, J. (2009). Sindrome di Tolosa Hunt: revisione basata su un caso clinico. Revistqa Memoriza.
- Díez de los Ríos González, A., Gómez Rebollo, C., e Aguilar Cuevas, R. (2013). Soluzione di caso 46. Sindrome di Tolosa-Hunt. radiologia.
- Granados-Reyes, G., Soriano-Redondo, E., e Durán-Ferreras, E. (2012). Sindrome di Tolosa-Hunt dopo un traumatismo oculare. Rev Neurol.
- Martínez, D., Casasco, J., Pendre, N., de Bonis, C., e Berner, S. (2010). TOLOSA-HUNT SYNDROME. Rev Argent Neuroc.
- NORD. (2016). Tolosa Hunt Syndrome. Estratto dalla National Organization for Rare Disorders.
- Sánchez Iñigo, L., e Navarro González, D. (2014). Sindrome di Tolosa-Hunt, un altro mal di testa. Neurol Arg.
- Taylor, D. (2015). Sindrome di Tolosa-Hunt. Ottenuto da Medscape.
- Zimmermann-Paiz, M. (2008). Sindrome di Tolosa-Hunt preceduta da paralisi facciale. Rapporto di un caso ... Rev Mex Oftalmol.